
Research
Our group wants to understand how mitochondria build and maintain their proteome. Our focus is on the role of mitochondrial proteases that are required for maturation and quality control of newly imported proteins. In particular, we are aiming to understand the pathophysiological consequences upon mutations and dysfunction in mitochondrial presequence proteases that result in severe neurodegeneration and cardiomyopathies. We investigate the consequences of imbalanced mitochondrial proteostasis and the protective cellular response that is triggered upon proteotoixc stress.
Mitochondria are essential eukaryotic organelles that are involved in a plethora of important cellular function like the ATP synthesis via oxidative phosphorylation, the synthesis of iron-sulfur-cluster, amino acids, heme or lipids or programmed cell death. To fulfill their diverse functions mitochondria are equipped with more than 1000 different proteins. The vast majority of these proteins are encoded in the nuclear DNA, translated on cytosolic ribosomes and imported into the organelle post-translationally. Targeting of these precursor proteins is mostly achieved by N-terminal extensions, termed presequences, that direct import across both mitochondrial membranes.
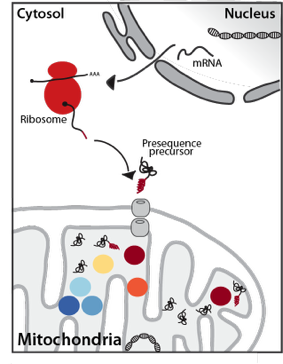
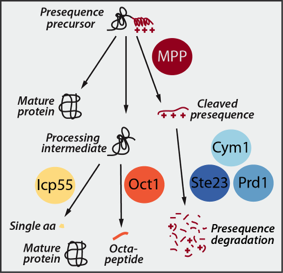
Upon import into the matrix the presequences are proteolytically removed and degraded by a network of presequence proteases. The essential mitochondrial processing protease MPP cleaves the presequence, which is subsequently degraded by the concerted action of three matrix peptidases: Cym1/PreP, Ste23/IDE and Prd1. While MPP can release mature, functional proteins, it often generates unstable intermediates. These processing intermediates are recognized and a second time processed by either the Intermediate cleaving peptidase Icp55 removing a single amino acid or by the octapeptidyl aminopeptidase Oct1/MIP, which cleaves off an octapeptide. This second processing reveals a stable N-terminal amino acid and increases the half-life of the protein.
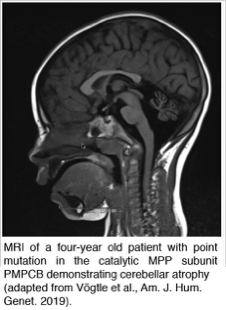
Dysfunctions in the mitochondrial presequence proteases result in severe human diseases like Alzheimer's disease, neurodegeneration or cardiomyopathies. We want to understand the pathological consequences caused by defective presequence proteases with their subsequent mitochondrial und cellular dysfunctions. For this we use the model organism Saccharomyces cerevisae, mice models, human tissue culture and patient cells.
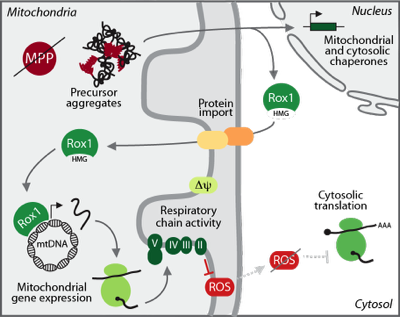
Disease mutations or other cellular stresses that result in mitochondrial imbalances are eliciting a global cellular response that triggers changes in nuclear transcription ("mitochondrial unfolded protein response"). We are investigating how the cell is sensing these imbalances and how mitochondria are molecularly remodeled to cope with and overcome stress in form of toxic protein aggregates. We have identified the nuclear transcription factor Rox1 to re-localize to mitochondria upon proteotoxic stress within the organelle, where it binds and maintains mitochondrial DNA to ensure translation of mitochondrial encoded proteins.
Nora Vögtle
Zentrum für Molekulare Biologie der Universität Heidelberg (ZMBH)
Im Neuenheimer Feld 282
D-69120 Heidelberg
n.voegtle(at)zmbh.uni-heidelberg.de